Amgen Worldwide
A biotechnology pioneer since 1980, Amgen has reached millions of patients around the world.
オンラインリソース
アムジェンは、患者さんが関心のある分野について学べるよう、多数のリソースを用意しております。
A biotechnology pioneer since 1980, Amgen has reached millions of patients around the world.
アムジェンは、患者さんが関心のある分野について学べるよう、多数のリソースを用意しております。
オテズラ錠10mg、20mg、30mg
オテズラの効能又は効果は、「局所療法で効果不十分な尋常性乾癬」、「乾癬性関節炎」および「局所療法で効果不十分なベーチェット病による口腔潰瘍」です[1]。
電子化された添付文書(電子添文)には以下の記載があります
4. 効能又は効果
○局所療法で効果不十分な尋常性乾癬
○乾癬性関節炎
○局所療法で効果不十分なベーチェット病による口腔潰瘍
5. 効能又は効果に関連する注意
〈局所療法で効果不十分な尋常性乾癬、乾癬性関節炎〉
5.1 以下のいずれかを満たす尋常性乾癬又は関節症性乾癬患者に投与すること。
・ステロイド外用剤等で十分な効果が得られず、皮疹が体表面積の10%以上に及ぶ患者
・難治性の皮疹又は関節症状を有する患者
1. オテズラ錠10mg, 20mg, 30mg 電子添文
※最新の情報は電子添文をご確認ください。電子添文のリンクはこちら
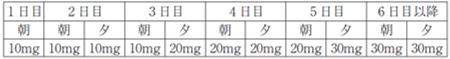
通常、成人にはアプレミラストとして以下のとおり経口投与し、6 日目以降はアプレミラストとして1 回30mgを1 日2 回、朝夕に経口投与します[1]。
漸増スケジュール
≪効能共通≫
・投与開始時に漸増投与を行わなかった場合、悪心、下痢、嘔吐等の発現率が高いことが示されているため、「用法及び用量」を遵守してください。
・重度の腎機能障害患者(Cockcroft-Gault式によるクレアチニンクリアランス値が30mL/min未満)では、本剤の血中濃度が上昇する可能性があることから、本剤を30mg 1 日1
回投与する等、減量も考慮し、慎重に投与してください。なお、本剤30mg 1 日1 回投与とする場合、投与開始時は朝の用量のみ投与してください。
≪局所療法で効果不十分な尋常性乾癬、乾癬性関節炎≫
・本剤による治療反応は、通常投与開始から24週以内に得られるため、24週以内に治療反応が得られない場合は、本剤の治療計画の継続を慎重に再考してください。
1. オテズラ錠10mg, 20mg, 30mg 添付文書
※最新の情報は電子化された添文文書(電子添文)をご確認ください。電子添文のリンクはこちら
オテズラ錠の吸収は食事の影響を受けません。そのため、投与については、朝、夕の1日2回以外の規定はございません。
<食事の影響の検討[1]>
健康な外国人の成人45例にアプレミラスト30mgを空腹時に投与及び高脂肪食(タンパク質、炭水化物及び脂肪からそれぞれ約150、250及び500~600カロリーを摂取)の開始30分後に投与したときのTmax(中央値)は、空腹時投与では2.5時間であり、食後投与では3.0時間でした。また、空腹時投与及び食後投与でのAUC及びCmaxはほぼ同等で、いずれも幾何平均値比%の90%信頼区間(CI)が80%~125%の範囲に含まれており、アプレミラストの吸収及び曝露に対する食事の影響は認められませんでした。
1. オテズラ錠10mg,20mg,30mgインタビューフォーム
※最新の情報は電子化された添付文書をご確認ください。電子添文のリンクはこちら
投与初日の朝に飲み忘れた場合
次の日の朝からスタートしてください。
投与2日目以降に飲み忘れた場合
気が付いた時に、1回分を飲んでください。ただし、次の飲む時間が近い場合は1回とばして、次の時間に1回分飲んでください。決して一度に2回分を飲まないでください。
1. オテズラ錠 患者向医薬品ガイド
※最新の情報は電子化された添付文書をご確認ください。電子添文のリンクはこちら
オテズラ錠の投与中止後の再投与について、漸増の規定はございません。
1. オテズラ錠10mg, 20mg, 30mg 添付文書
※最新の情報は電子化された添付文書をご確認ください。電子添文のリンクはこちら
電子化された添付文書(電子添文)において、『錠剤を噛み砕いたり、割ったりしないこと。』と注意喚起しております。
また、錠剤に割線はなく、分割や粉砕後の安定性、有効性、安全性、薬物動態のデータはございません。
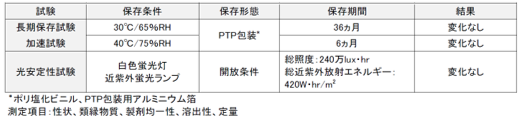
<有効成分の吸湿性および製剤の安定性データ>
●有効成分
吸湿性:相対湿度0~95%で吸湿性は認められなかった
●製剤
長期保存試験:30℃/65%RH、PTP包装、36ヵ月保存した結果、変化なし
加速試験:40℃/75%RH、PTP包装、6ヵ月保存した結果、変化なし
光安定性試験:白色蛍光灯、開放条件で総照度240万lux・hr及び、近紫外蛍光ランプ、開放条件で総近紫外放射エネルギー420W・h/m2 曝光した結果、変化なし
1. オテズラ錠10mg, 20mg,30mgインタビューフォーム
※最新の情報は電子添文をご確認ください。電子添文のリンクはこちら
一包化後の安定性を検討していないため、不明です。
投与開始時のスターターパックについては誤投与を避けるため、一包化は推奨しておりません。
※最新の情報は電子化された添付文書をご確認ください。電子添文のリンクはこちら
簡易懸濁後の安定性および有効性、安全性の検討は行われていないため、簡易懸濁の可否は不明です。
※最新の情報は電子化された添付文書をご確認ください。電子添文のリンクはこちら
製剤の長期保存試験、加速試験、光安定性試験の結果、何れの測定項目も変化を認めませんでした。
1. オテズラ錠10mg, 20mg, 30mgインタビューフォーム
※最新の情報は電子化された添付文書をご確認ください。電子添文のリンクはこちら
一般に高齢者では生理機能が低下しているため、感染症、下痢、悪心、嘔吐等の副作用の発現に留意し、患者の状態を十分に観察しながら、慎重に投与してください。
電子化された添付文書(電子添文)には以下の記載があります。
9. 特定の背景を有する患者に関する注意
9.8 高齢者
感染症、下痢、悪心、嘔吐等の副作用の発現に留意し、患者の状態を十分に観察しながら、慎重に投与すること。一般に生理機能が低下している。
1. オテズラ錠10mg, 20mg, 30mg 添付文書
※最新の情報は電子添文をご確認ください。電子添文のリンクはこちら
小児等は臨床試験では除外されています。
本剤の小児等における安全性は確立しておりません。
添付文書(電子添文)には以下の記載があります。
9. 特定の背景を有する患者に関する注意
9.7 小児等
小児等は臨床試験では除外されている。
1. オテズラ錠10mg, 20mg, 30mg 添付文書
※最新の情報は電子化された添付文書をご確認ください。電子添文のリンクはこちら
オテズラ錠の薬効成分であるアプレミラストは、PDE4を阻害する低分子の経口PDE4阻害剤で、細胞内で炎症性及び抗炎症メディエーターのネットワークを調節します。PDE4はcAMPに特異的なPDEで、主に炎症性細胞に分布しています。本剤は、PDE4を阻害することにより細胞内cAMP濃度を上昇させ、IL-17、TNF-α、IL-23、及び他の炎症性サイトカインの発現を制御することにより炎症反応を抑制します。
<薬理作用>
・In vitroにおける薬理活性
1.
cAMPの加水分解により測定したホスホジエステラーゼ4(PDE4)活性に対する競合的かつ可逆的な阻害作用を示しました(IC50=74nM、Ki=68nM)。また、PDE4A、PDE4B、PDE4C、PDE4Dのいずれのサブタイプに対しても阻害作用を示しました。
2. ヒト由来精製T細胞において、IL-17等の炎症性サイトカインの産生抑制作用を示しました(IL-17産生抑制:IC50=90nM)。
3. ヒト末梢血単核球細胞において、TNF-α等のエンドトキシン誘発性の炎症性サイトカインの産生抑制作用を示しました(TNF-α産生抑制:IC50=110nM)。一方、抗炎症サイトカインであるIL-10の産生増加作用を示しました。
・In vivoにおける薬理活性
1. ヒト皮膚/乾癬NK細胞を異種移植したBeige-重症複合免疫不全マウスモデルにおいて、アプレミラスト(
5mg/kg/day)は表皮の異常肥厚・増生、乾癬病変所見、病変組織におけるTNF-α、
ヒト白血球抗原-DR(HLA-DR)、細胞間接着分子-1(ICAM-1)の発現を抑制しました。
2. 抗Ⅱ型コラーゲンモノクロナール抗体やⅡ型コラーゲン免疫により作成されたマウスの関節炎モデルにおいて、アプレミラスト( 5mg/kg/day及び25mg/kg/day)は症状スコアを抑制しました。
1. オテズラ錠10mg, 20mg, 30mgインタビューフォーム
※最新の情報は電子添文をご確認ください。電子添文のリンクはこちら
オテズラ錠スターターパック:27錠((10mg× 4 錠、20mg× 4 錠、30mg×19錠)× 1 パック)とオテズラ錠30mg:56錠(14錠(PTP)× 4 シート)の2規格です。
1. オテズラ錠10mg, 20mg, 30mg 電子化された添付文書
※最新の情報は電子添文をご確認ください。電子添文のリンクはこちら
本剤の投与は適応疾患である尋常性乾癬、乾癬性関節炎およびベーチェット病の治療に十分な知識・経験を持つ医師のもとで行ってください。
1. オテズラ錠10mg, 20mg, 30mg 電子化された添付文書
※最新の情報は電子添文をご確認ください。電子添文のリンクはこちら
肝機能障害患者へのオテズラ錠投与について、減量の規定はございません。
中等度(Child-Pugh 7~9
)又は重度(Child-Pugh10~13)の肝機能障害患者においてアプレミラストとその主要代謝物の薬物動態について、影響は認められませんでした。
1. オテズラ錠10mg, 20mg, 30mg 電子化された添付文書
※最新の情報は電子添付をご確認ください。電子添文のリンクはこちら
妊産婦、授乳婦に対する安全性は確立していません。アプレミラストの添付文書(電子添文)では、妊婦又は妊娠している可能性のある女性は禁忌に該当します[1]。
電子化された添付文書(電子添文)の「禁忌」、「妊婦、産婦、授乳婦等への投与」の項では、以下の内容が記載されています。
2. 禁忌(次の患者には投与しないこと)
2.2 妊婦又は妊娠している可能性のある女性
9. 特定の背景を有する患者に関する注意
9.5 妊婦
妊婦又は妊娠している可能性のある女性には投与しないこと。マウスで臨床用量の2.3倍に相当する用量で早期吸収胚数及び着床後胚損失率の増加、胎児体重の減少、骨化遅延が、サルで臨床用量の2.1倍に相当する用量で流産が認められており、ヒトにおいて胚胎児毒性を引き起こす可能性が否定できない。[2.2、9.4参照]
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。本剤のヒトにおける乳汁への移行は不明であるが、本剤を投与した動物試験(マウス)で乳汁への移行が報告されている。
1. オテズラ錠10mg, 20mg, 30mg 電子添文
※最新の情報は電子添文をご確認ください。電子添文のリンクはこちら
オテズラ錠のオーストラリア医薬品評価委員会基準はCategory B3です。
1.オテズラ錠10mg,20mg,30mgインタビューフォーム
※最新の情報は電子化された添付文書をご確認ください。電子添文のリンクはこちら
弊社医療関係者向けホームページをご確認ください。
https://www.amgen.co.jp/products/for-physicians
製品の詳細および最新の情報は添付文書をご参照ください。
弊社製品の使用後に有害事象が認められた場合、弊社製品に不具合が認められた場合は、弊社担当者までご報告をお願いいたします。引き続き、弊社製品の詳細調査などへのご協力お願いいたします。

弊社製品情報に関するご質問への回答や、関連する選択肢が表示され、お求めの情報に速やかに到達することができます。

医療関係者を対象とした、製品に関するお問い合わせ窓口です。
平日9:00~17:30 (土日・祝日・会社休日を除く)の間、受付しております。
アムジェン株式会社では、お問い合わせ内容を正確に聞き取るため、また回答の質の維持・向上のため、当センターにおける受発信の通話を録音させていただいております。